Scientific Tools
The Novo Nordisk Foundation Center for Biosustainability has developed several molecular and bioinformatic tools, which can help life scientists improve cell performance. The tools can accelerate and aid the design, monitoring and optimisation of cell response to different genomic changes. Furthermore, we offer engineered cell lines upon request.
Software tools, platforms and models
antiSMASH
The antibiotics and Secondary Metabolites Analysis SHell (antiSMASH) is a fully automated pipeline to mine bacterial and fungal genome data for secondary metabolite biosynthetic gene clusters.
The small molecules encoded by these biosynthetic gene clusters often have various bioactivities including anti-microbial, anti-cancer, anthelmintic and others. Therefore, they are lead compounds for many drugs like antibiotics.
antiSMASH is the most widely used tool for these genome mining approaches. Since its initial release in 2011, more than 160,000 analyses have been performed on the public antiSMASH service, which recently was moved to DTU Biosustain.
References:
- Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H.U., Bruccoleri, R., Lee, S.Y., Fischbach, M.A., Müller, R., Wohlleben, W., Breitling, R., Takano, E., Medema, M.H., (2015) antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237-W243.
- Blin, K., Kazempour, D., Wohlleben, W., Weber, T., (2014) Improved lanthipeptide detection and prediction for antiSMASH. PLoS ONE 9, e89420. (Open Access)
- Blin, K., Medema, M.H., Kazempour, D., Fischbach, M., Breitling, R., Takano, E., Weber, T., (2013) antiSMASH 2.0 – a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204-W212. (Open Access)
- Medema, M.H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M.A., Weber, T., Takano, E., Breitling, R., (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339-W346. (Open Access)
AntiSmash links
Contact
Tilmann Weber Professor tiwe@dtu.dk
Cameo
Cameo is a high-level python library developed to aid the strain design process in metabolic engineering projects.
The Cameo library provides a modular framework of simulation methods, strain design methods and access to models which target developers that want custom analysis workflows. Furthermore, it exposes a high-level programming interface to users that simply want to compute promising strain designs.
Cameo links
iCHO1766
iCHO1766 is a model of the biosynthetic pathways in CHO cells, and includes 6643 metabolic reactions and 2341 metabolites.
iCHO1766 links
COBRApy
COBRApy is a package for constraint-based modelling of metabolic networks written in Python.
The COBRApy tool strives to become the reference software library for doing genome-scale metabolic modelling by providing the essentials (data structures, simulation methods and model import/export) that others can base their more specialised software tools on.
COBRApy links
CRISPy-web
CRISPy-web is an online tool to design single guide RNAs (sgRNAs) for CRISPR applications with microbes. Unlike most other CRISPR design tools, CRISPy-web allows the user to upload custom/non-model organisms genome data to screen for off-target effects.
CRISPy-web can directly access antiSMASH results and use the antiSMASH-annotation to aid with the sgRNA design. The initial version of CRISPy-web was based on CRISPy, a web-tool to find sgRNAs for Chinese hamster ovary (CHO-K1) cells.
References
- Blin, K., Pedersen, L.E., Weber, T., Lee, S.Y., (2016) CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synth. Syst. Biotechnol., in press, doi: 10.1016/j.synbio.2016.1001.1003. (Open Access)
- Ronda, C., Pedersen, L.E., Hansen, H.G., Kallehauge, T.B., Betenbaugh, M.J., Nielsen, A.T., Kildegaard, H.F., (2014) Accelerating genome editing in CHO cells using CRISPR Cas9 and CRISPy, a web-based target finding tool. Biotechnol. Bioeng. 111, 1604-1616.
CRISPy-web links
Contact
Tilmann Weber Professor tiwe@dtu.dk
Kai Blin Senior Researcher kblin@dtu.dk
MASS Toolbox
The Mass-Action Stoichiometric Simulation (MASS) toolbox is a modelling software package that focuses on the construction and analysis of kinetic and constraint-based models of biochemical reactions systems.
It has been developed in the Section for Network Reconstructions and in Silico Biology at the University of California, San Diego, and is now jointly maintained by UCSD and DTU Biosustain.
MASS Toolbox links
Second Metabolite Bioinformatics Portal
The Secondary Metabolite Bioinformatics Portal, coordinated by the New Bioactive Compound Section, is a community-driven online resource providing a comprehensive catalogue of tools for Omics-based studies of microbial secondary metabolite biosynthetic pathways.
The tool provides short descriptions of specialised web servers, stand-alone tools and databases, provides links to the respective web-sites and literature references of the different tools.
Reference
- Weber, T., Kim, H.U., (2016) The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Synth. Syst. Biotechnol., in press, doi: 10.1016/j.synbio.2015.1012.1002. (Open Access)
Second Metabolite Bioinformatics Portal links
Contact
Tilmann Weber Professor tiwe@dtu.dk
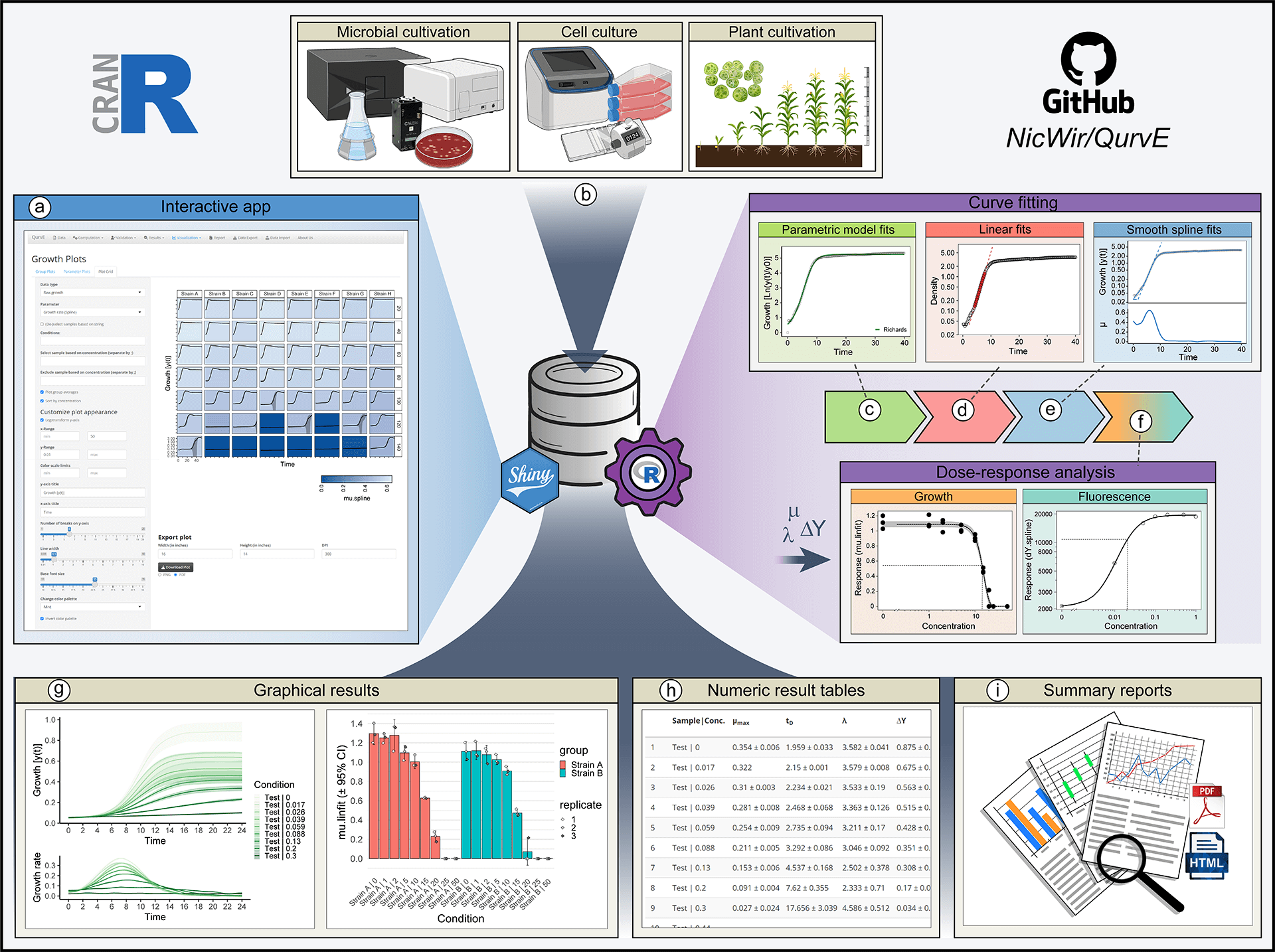
QurvE
Advanced analysis and visualization of growth and fluorescence curves.
QurvE application
QurvE R-package
View larger version of the figure
{kind=link}
Figure: QurvE enables robust, high-throughput analysis of growth and fluorescence data. (a) All functionalities within QurvE are accessible via an intuitive graphical user interface created with shiny, which can be installed locally on Windows PCs. (b) Any type of biological growth data can be analyzed. For commonly used cultivation devices, a growing list of data parser functions allows the conversion of exported experimental data into the QurvE-compatible format. In a single computation workflow, three different types of algorithms are performed on every sample in a dataset: (c) Five parametric models are fit to the data to find an equation that best describes the growth curve. (d) Relevant (log-) linear phases are extracted from each sample to perform robust linear regression. (e) The representation of data points with cubic smoothing splines allows extraction of growth rates over time and applies to any curve shape. (f) Relevant parameters (growth rates, biomass yields, rate of fluorescence increase, etc.) can be used in combination with concentration data to analyze dose-response relationships. This is done by applying either dose-response models or smoothing splines. (g) Dedicated plot functions facilitate the fit validation, the interpretation of results, and, due to the availability of numerous customization options, the generation of suitable for publication. (h) All computed parameters can be exported as table files or inspected interactively from within the app. (i) All chosen fitting options as well as numerical and graphical results can be compiled in reports in PDF and HTML format to promote data transparency and good scientific practice. In this spirit, raw data and results can be exported as a single data container in the form of an .RData file to give other researchers access to data and analysis methods used.
Contact
Pablo Ivan Nikel Professor & Group Leader pabnik@dtu.dk
Memote
Memote is a community-maintained, standardised set of metabolic model tests.
The Memote tests cover a range of aspects from annotations to conceptual integrity and can be extended to include experimental datasets for automatic model validation.
Memote links
Vectors, plasmids and strains
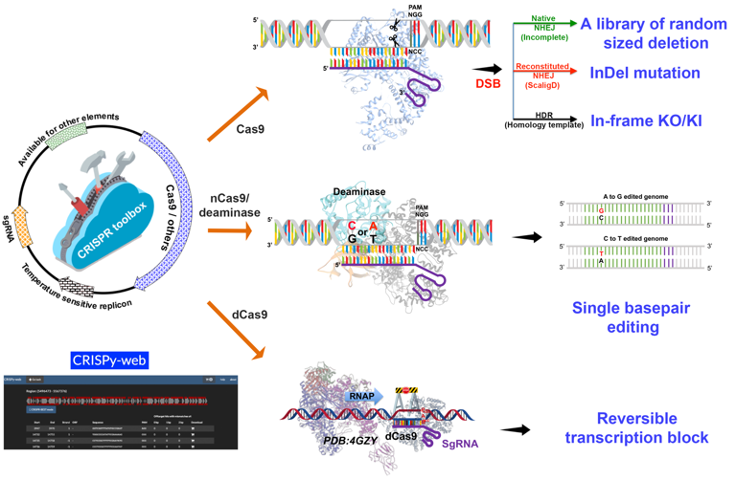
CRISPR toolkit for Actinomyces
The CRISPR toolkit for Actinomyces includes both classic CRISPR-Cas9 related and CRISPR base editing based genetic manipulation tools for Actinomyces.
All plasmids are avaliable at Addgene. Furthermore, the toolkit is complemented by the sgRNA design software CRISPy-web.
pCRISPR-Cas9 (Addgene: 125686) can be used to generate deletion libraries by a single transformation; or to generate seamless deletion/insertion by providing editing templates.
pCRISPR-Cas9-ScaligD (Addgene: 125688) can be used to introduce small InDels.
pCRISPR-dCas9 (Addgene: 125687), CRISPRi, can be used to modulate transcriptional activity of target genes.
pCRISPR-cBEST (Addgene: 125689) and pCRISPR-aBEST (Addgene: 131464) can be used to convert C:G basepairs to T:A basepairs and A:T basepairs to G:C basepairs, respectively.
Contact
Tilmann Weber Professor tiwe@dtu.dk
Genome editing tools for yeasts
EasyClone ease the integration of multiple genes simultaneously – with or without markers.
EasyClone vector set for simultaneous integration of multiple genes into Saccharomyces cerevisiae genome with an option of recycling selection markers. Available from BCCM Collection. [Order] [Jensen et al, 2014]
EasyClone 2.0 vector set with dominant selection markers for Saccharomyces cerevisiae. Available on AddGene. [Order] [Manual] [Stovicek et al, 2015]
EasyClone-MarkerFree vector set for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Available on AddGene. [Order] [Manual] [Jessop-Fabre & Jakočiūnas et al, 2016]
EasyClone-MarkerFree expansion. Available on AddGene. [Order] [Babaei et al., 2021]
EasyCloneMulti vector set for simultaneous and multiple genomic integrations in Saccharomyces cerevisiae. Available from AddGene. [Order] [Maury et al, 2016]
EasyCloneYALI vectors for CRISPR and marker-mediated engineering of Yarrowia lipolytica. Available on AddGene. [Order] [Manuals] [Holkenbrink et al., 2018]
Transportome engineering vector libraries allow knock-out of 361 non-essential transporters in Saccharomyces cerevisiae via CRISPR-Cas9. Available on AddGene. [Order] [Wang et al., 2021]
Contact
Irina Borodina Professor irbo@dtu.dk
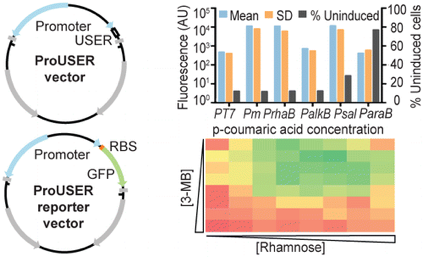
ProUSER vectors
The ProUSER vector collection is a set of vectors that facilitates fast and efficient USER cloning, without the need for PCR amplifying the backbones or promoter sequences.
References
- Calero P, Jensen SI, Nielsen AT (2016) Broad-host-range ProUSER vectors enable fast characterization of inducible promoters and optimization of p-coumaric acid production in Pseudomonas putida KT2440. ACS Synt. Biol. 5:741-753
Contact
Alex Toftgaard Nielsen Professor altof@dtu.dk
Sheila Ingemann Jensen Senior Researcher shin@dtu.dk
Seven-In-Seven
Seven-in-Seven is a newly developed method for fast generation of multiplex gene deletions in E. coli.
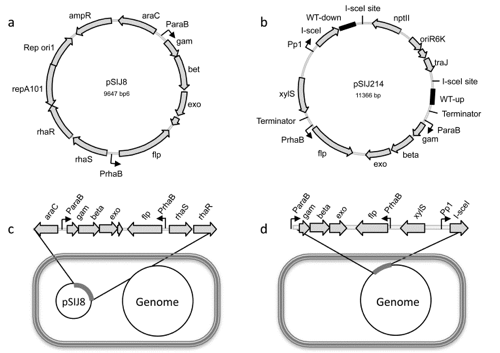
References
- Jensen SI, Lennen R, Herrgård MJ, Nielsen AT (2015). Seven gene deletions in seven days: Fast generation of Escherichia coli strains tolerant to acetate and osmotic stress. Scientific Reports 5: 17874. doi: 10.1038/srep17874
Contact
Alex Toftgaard Nielsen Professor altof@dtu.dk
Sheila Ingemann Jensen Senior Researcher shin@dtu.dk
Engineered Saccharomyces cerevisiae for production of chemicals
All the published strains are available upon request.
- For production of 3-hydroxypropionic acid from glucose via malonyl-CoA pathway [Kildegaard & Jensen et al, 2016]
- For production of 3-hydroxypropionic acid from xylose [Kildegaard et al, 2015]
- Industrial xylose-utilizing S. cerevisiae strains [Stovicek et al, 2015]
- For production of resveratrol [Li et al, 2015]
- For production of p-coumaric acid [Rodriguez et al, 2015]
- For production of 3-hydroxypropionic acid from glucose via beta-alanine pathway [Borodina et al, 2015]
- Tolerant to 3-hydroxypropionic acid [Kildegaard et al, 2014]
Contact
Irina Borodina Professor irbo@dtu.dk
pFREE
A versatile one-step CRISPR-Cas9 based approach to plasmid-curing
The pFREE system is a CRISPR-Cas9 based platform that offers plasmid curing of all major classes of vectors used for cloning and expression purposes in bacteria. The temperature-sensitive and broad host-range version of pFREE (pFREE-RK2) was also developed to provide a curing solution for broad range of Gram-negative bacteria.
The curing workflow is performed in a fast one-step protocol that only requires transformation of pFREE into the strain harboring the target plasmids for curing and the addition of the pFREE inducers, followed by overnight incubation. The pFREE plasmid holds a self-curing feature that allows for efficient and complete plasmid-curing.
All the different versions of pFREE and pFREE-RK2 are available from Addgene.
Reference
- Lauritsen, I*., Porse, A*., Sommer, M. O. A., & N rholm, M. H. H. (2017). A versatile one-step CRISPR-Cas9 based approach to plasmid-curing. Microbial Cell Factories, 16, [135]. https://doi.org/10.1186/s12934-017-0748-z